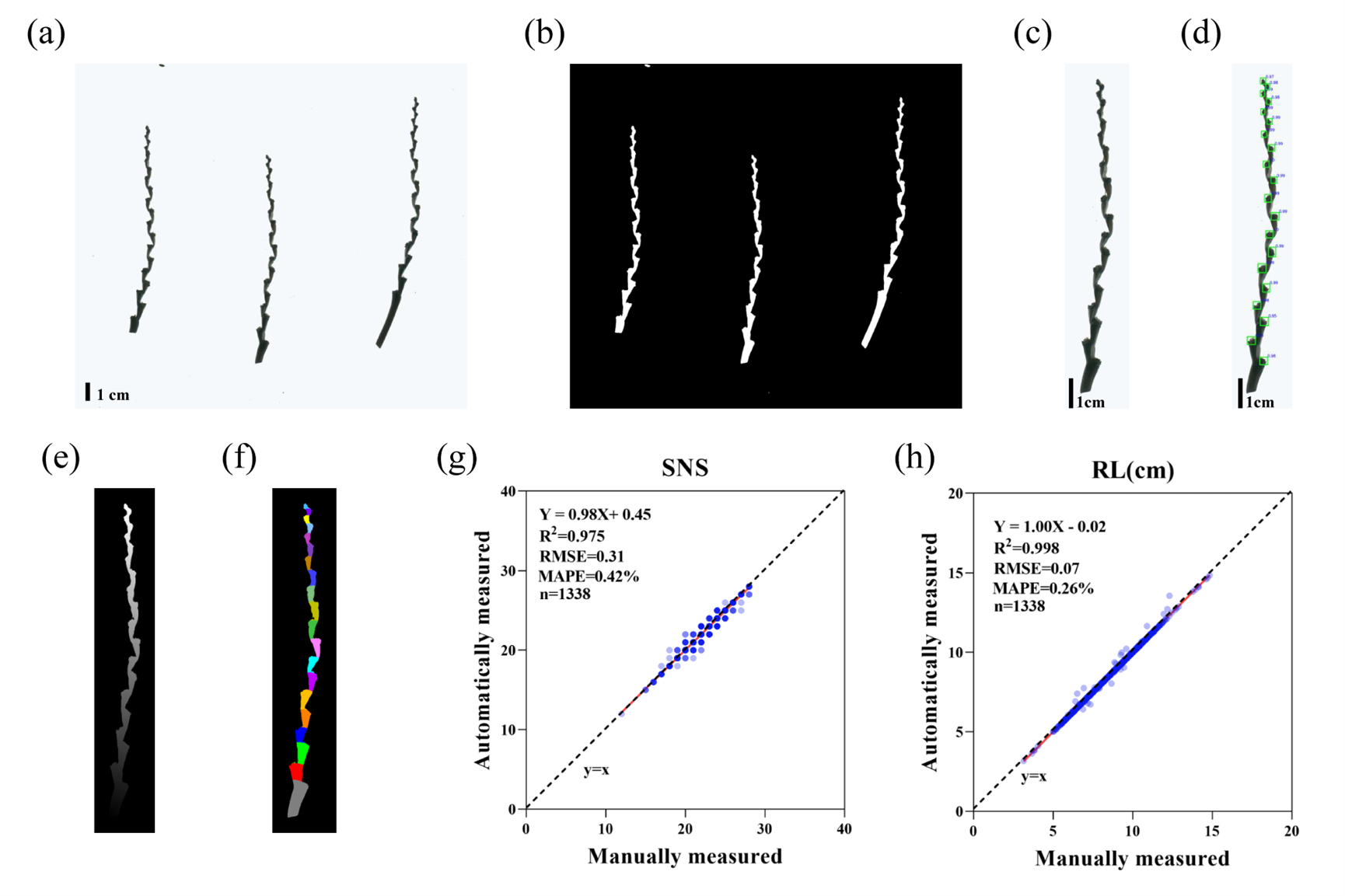
梁承志研究组构建出一个目前最高质量的植物基因组参考序列
发布时间:2017.05.05
随着PacBio单分子实时(SMRT)测序技术的发展,利用它已能够独立完成高质量基因组草图的组装。然而,这些草图序列中仍然存在多种错误,比如序列中会包含有很多嵌合体(即不同位置的序列连接到了一起)或是组装质量比较差的区域等,特别是重复序列区域有的没有组装出来,有的组装出来多个序列,而且这些错误通常也难以检测。另外,单纯利用SMRT测序还只能装出一个个的DNA小片段,而要连成整条染色体的序列,则需要借助于遗传图谱,或是最近发展的Hi-C技术。然而,简单利用于这两个技术形成的染色体序列,仍存在很多的缺陷:(1)小的片段难以放到染色体上,导致形成的染色体序列上有大量的缺口,(2)放上的片段中包含有大量的顺序排列错误和序列的方向错误。这样形成的染色体用作参考基因组进行基因定位,容易漏掉基因或导致错误的定位。
为了利用现有的技术进行高质量的植物基因组组装,中国科学院遗传与发育生物学研究所梁承志研究组与四川农业大学李仕贵教授合作,从2014年开始对一个籼稻基因组蜀恢498(R498)进行PacBio单分子测序,结合遗传图谱和fosmid文库测序,并利用了BioNano光学图谱的验证,最后得到一个长度为390.3 Mb的基因组,共由17个连续DNA片段(Super-Contig)组成,包括7条头尾相连的染色体和5条分成两个Super-Contig的染色体。蜀恢498的基因组是目前所有高等动植物中组装质量最高的基因组,除了5个着丝粒重复序列区域和其它少数几个串联重复序列区域,整个基因组都被组装了出来;其基因组完整性和连续性都大大高于日本晴及拟南芥等基因组,且有更低的错误率。这个结果也显示了籼稻的基因组大小不超过395 Mb。他们在R498序列中发现了两个核仁组织区,多于日本晴基因组的一个。通过比较两个基因组上的基因序列可以看出,超过2/3的基因有序列上的差异,两个基因组之间也含有大量的由于转座子独立插入导致的结构变异。此外,他们还组装出了一个完整的线粒体序列,发现了日本晴线粒体序列中的几个大的错误,也发现了目前日本晴基因组参考序列中错误地掺入了很多线粒体和叶绿体的序列。作为参考基因组,R498序列将被用于籼稻突变基因的定位,及籼稻群体的全基因组关联分析。蜀恢498基因组的完成,显示了在现有的技术条件下,得到一个接近完成并在染色体水平上具有连续序列的高质量参考基因组是可行的,对于提高目前高等动植物基因组的组装质量具有重要的指导意义。
该项研究于2017年5月4日在Nature Communications杂志在线发表(DOI:10.1038/ncomms15324)。梁承志研究组博士生杜会龙,工作人员于莹博士、马延飞博士、高强和曹英豪为该论文的共同第一作者。该文通讯作者是遗传发育所的梁承志研究员和四川农业大学水稻研究所的李仕贵教授。该研究得到了中国科学院战略性先导专项和国家自然科学基金项目的资助。
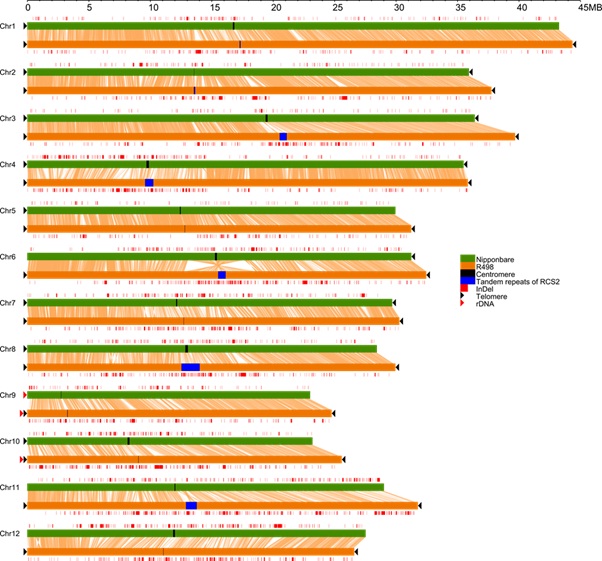
图1:蜀恢498和日本晴全基因的比较显示了染色体端粒的有无及二者之间的结构变异分布。