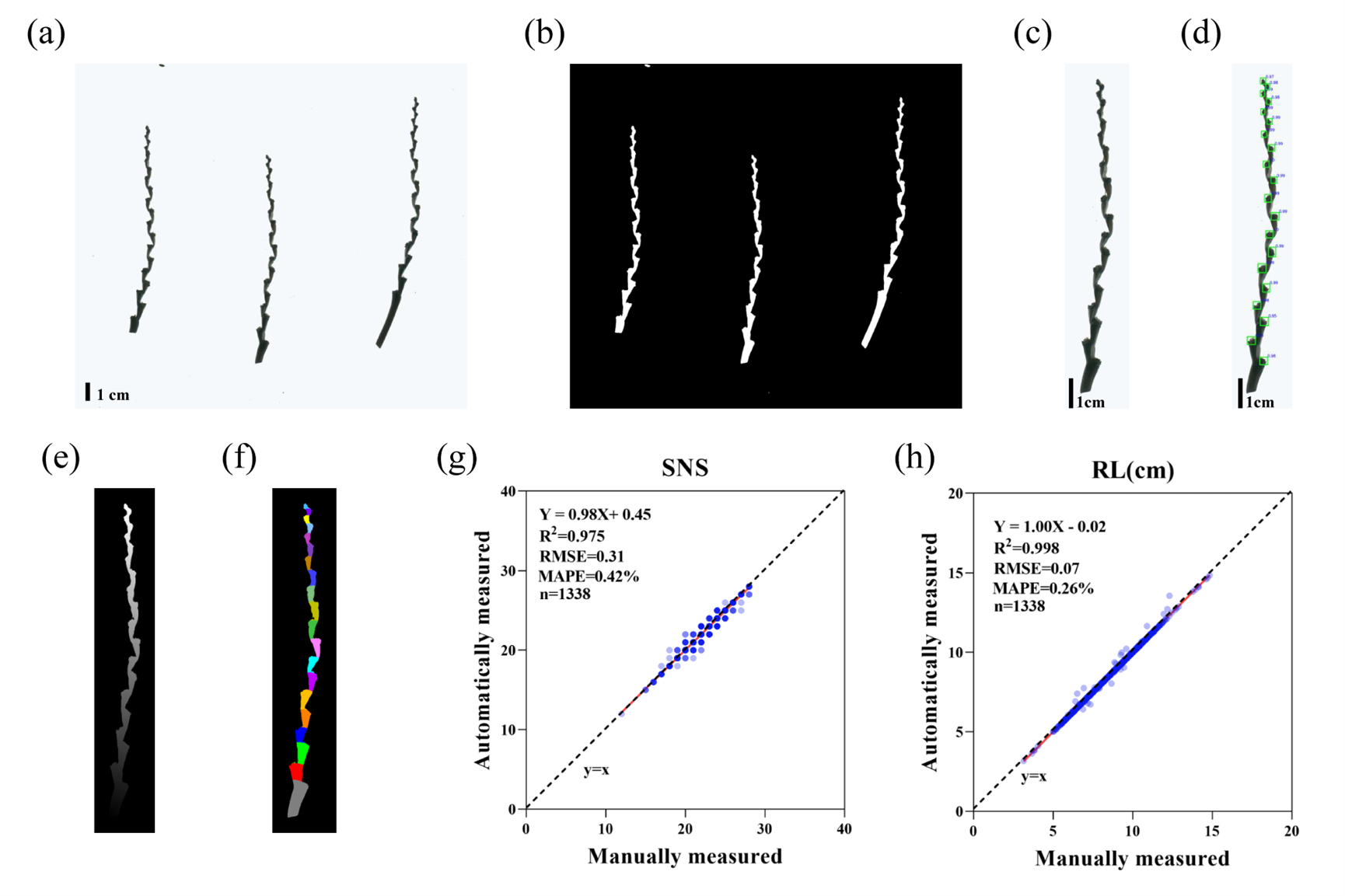
鲁非研究组开发高效率群体转录组研究技术
发布时间:2021.09.16
RNA测序(RNA-Seq)是现代生物学研究的基石技术之一,将许多研究从单一的基因组水平提升到多组学水平,是解析基因组功能的强大工具。现有的全基因组水平的大规模测序以及高质量泛基因组的组装,为大规模转录组研究提供了广阔的空间,迫切需要高效的RNA-Seq技术进行深层次生物学解析。本研究综合了已报道的3' RNA-Seq定制化建库方法的优点,并针对测序外包市场标准双端150bp/250bp(PE150/250)模式优化实验设计,开发了一种通用高效的基因表达文库构建方法,即简化的poly(A)锚定测序(SiPAS)技术。通过在六倍体面包小麦中SiPAS性能的测试实验,证实了SiPAS具有高水平的灵敏度、准确性和可重复性,该技术有望成为群体转录组学解析的利器,极大促进作物和许多其他植物遗传学研究。
本研究基于已有的3' RNA-Seq的优势和标准的PE150测序模式,从以下三个方面进行改进并评估。1)优化建库流程更适用标准的PE150测序模式,因为更长的读段可以显著提高比对的准确性。2)由于碱基质量对比对的准确性有积极影响,通过调转测序接头,使用在Illumina测序中有着更高测序质量的R1端进行非poly(T)端的测序,提高测序质量。3)单细胞RNA-seq常用特异分子标识符(UMI)来校正PCR扩增导致的基因表达定量偏差,本研究评估了UMI对bulk RNA-seq的校正效果。因此,设计了四种流程测试,即T1,T2(调转接头),T3(添加UMI),T4(调转接头并添加UMI)进行对比实验(图1)。
本研究首先使用了模拟数据证实长读段和高碱基质量可以显著提高序列比对的灵敏度(从0.75提高至0.95)。在实验结果中,通过调转接头提高碱基质量可以使单端比对的唯一比对率增加10.37%。在单细胞中常用UMI矫正PCR扩增偏好性,由于bulk RNA-Seq建库的RNA起始量高扩增循环数少,UMI对bulk RNA-Seq并非必需。在基因表达定量方面,长读段和高碱基质量也可以显著提高检测准确性和重复性。综合上述结果,优选出T2(调转接头)建立SiPAS技术。
优选后的SiPAS在基因差异表达分析方面有着与TruSeq类似的高效检测能力,PCA分析表明,代表生物学差异的PC1解释了总方差的78%,而代表SiPAS和TruSeq之间技术差异的PC2仅占总差异的18%。同时,两种建库方法对DEG (Differentially expressed gene) 鉴定的一致性达到了0.95(图2)。另外,在使用Mg++离子随机片段化模拟RNA降解的实验中,SiPAS对降解RNA具备良好的检测能力,与对RNA完整性要求非常严格的建库方法相比,SiPAS技术更适用于高通量群体转录组研究。
综上,SiPAS在实现经济高效($2/样本)的基础上,在基因表达定量方面达到了与TruSeq类似的性能,SiPAS有望在大规模群体转录组研究中发挥重要作用,成为基因组功能解析的利器。
该研究成果于2021年9月12日在线发表于Plant Biotechnology Journal杂志上(DOI:10.1111/pbi.13706),其核心技术已经申请国家发明专利(申请号:202111058665.4)。中国科学院遗传与发育生物学研究所鲁非组工程师王静、博士研究生徐俊和杨晓寒为该研究的共同第一作者,鲁非研究员为该研究的通讯作者。研究得到中国科学院战略性先导科技专项种子精准设计与创造(XDA24020201和XDA24040102)和国家自然科学基金(31970631和31921005)的支持。
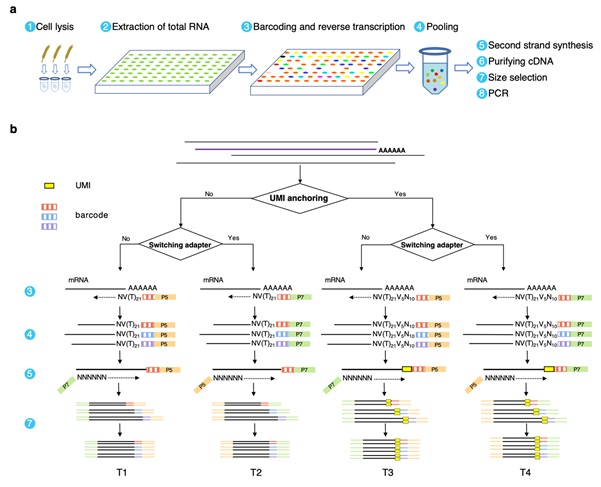
图1:SiPAS四种测试流程设计原理图
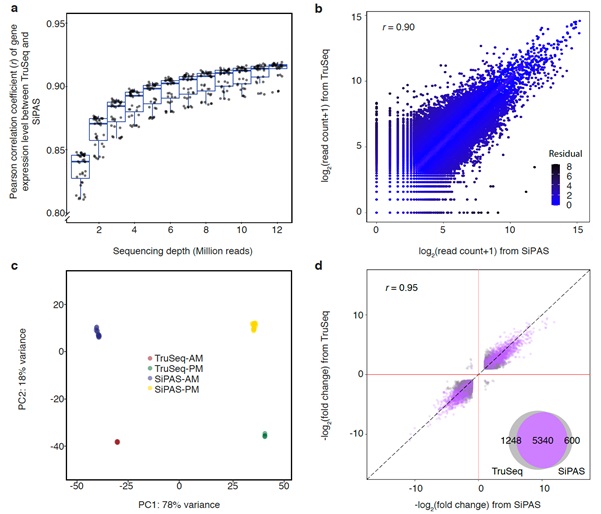
图2:SiPAS与TruSeq的性能比较